SeqAn Nodes for KNIME
SeqAn is an open source C++ library of efficient algorithms and data structures for the analysis of sequences with the focus on biological data. SeqAn is free software available under the three clause BSD license and runs under Windows, Mac OS X and Linux. The library applies a unique generic design that guarantees high performance, generality, extensibility, and integration with other libraries. SeqAn is easy to use and simplifies the development of new software tools with a minimal loss of performance.
The software library is intended to allow rapid prototyping of algorithms for analyzing large sets of sequences. The main emphasis lies on NGS data. SeqAn supports the handling of large sets of reads. It is in worldwide use for developing analysis tools for genomic sequence analysis.
The main contributor of SeqAn is currently the Algorithmic Bioinformatics Group at the Freie Universität Berlin. For a more detailed description we refer you to the SeqAn book published at CRC press and available from all bookstores.
Source Code
The source code of SeqAn is hosted at GitHub and can be accessed at GitHub. Note that GitHub hosts the sources of the SeqAn library and tools, the actual Java sources of KNIME nodes is automatically generated by GKN. If you want to develop your own KNIME nodes with SeqAn.
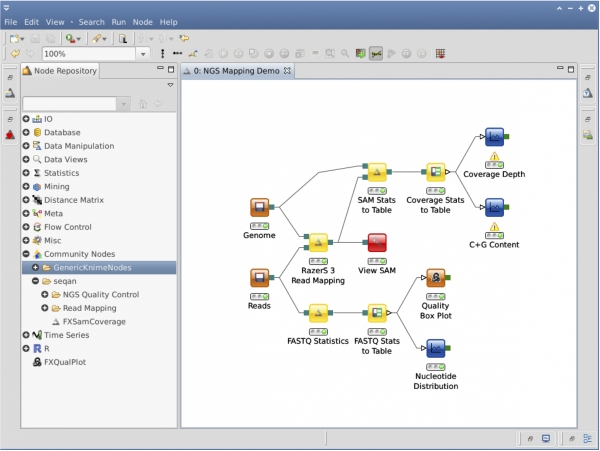
NGS Workflows
Please read KNIME HowTo about how to import SeqAn nodes in KNIME and how to develop your own nodes. If you want to contribute a whole workflow to the SeqAn community or use workflows from others you can do that on:
https://github.com/seqan/knime_seqan_workflows
Fork the git repository into your own repo and clone it to your local machine. To add a new workflow, create a folder WORKFLOWNAME_workflow. From within KNIME export your workflow without the data files as a .zip file into that folder. Provide a README, a screenshot and some examples. After everything is ready, commit the folder to your repo and create a github pull request.