What is virtual screening in pharmaceutical R&D?
Drug discovery projects are long R&D processes that last more than 10 years to reach the patient and have a high risk of failure. In small molecule research, the goal of these projects is to identify those chemical structures that interact with key receptors, have drug-like properties, and that are not already known. In this context, finding good starting points is critical in any drug discovery project. These starting points can come from extensive experimental testing, which is a costly and long process, or might rely on the use of computers to speed up the process. In a virtual screening, computers are used to analyze libraries of millions of compounds to identify which ones are more promising.
PharmScreen for KNIME
PharmScreen for KNIME is a set of nodes from Pharmacelera, oriented to help chemists in their drug discovery projects to find leads with higher chances of becoming a drug. PharmScreen nodes find candidate molecules with greater chemical diversity by searching proprietary, public or commercial compound libraries.
Both nodes are parallelized to take advantage of all the computing power of your PC, workstation, or cluster without having to go through any configuration hurdles.
Main features
Pharmacelera’s nodes enable you to perform a variety of tasks such as:
- Increase the chemical diversity of your candidate molecules
- Enrich your compound library
- Find alternative scaffolds not covered by existing IP
- Overcome pharmacological limitations of your hits
- Evaluate the selectivity of your hits for target / anti-target
- Repurpose your candidate molecules for other therapeutic areas
What is the underlying science?
PharmScreen uses a unique and superior 3D representation of molecules based on electrostatic, steric, and hydrophobic interaction fields derived from semi-empirical Quantum-Mechanics (QM) calculations. Such fields describe with high accuracy the factors that determine ligand / receptor interactions. These chemo-type agnostic descriptors enable identification of the compounds with similar physico-chemical properties but with different and diverse molecular scaffolds.
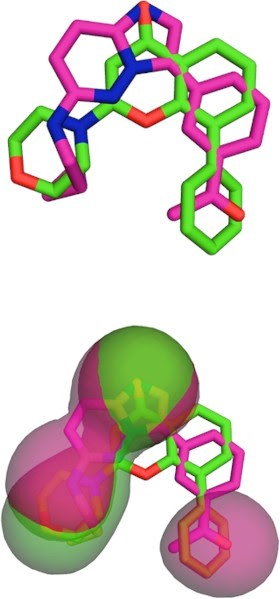
Molecular recognition is a central biochemical process. It defines drug interaction with biomolecules. This recognition is largely driven by hydrophobicity: hydrophobic areas of drug compounds tend to match hydrophobic areas of binding sites and cavities of macromolecules.
Hydrophobicity is often neglected in existing in-silico tools, which tend to focus their algorithms on electrostatic, hydrogen bonds, and steric components. As a consequence, chemical space is not properly mined and the proposed new chemical structures tend to be constrained and repetitive.
PharmScreen for KNIME offers a robust solution to this problem based on new molecular hydrophobicity descriptors. These differential descriptors overcome the above-mentioned drawbacks and lead to clear improvements. Precisely, more complete and original description of chemical space is achieved which enables finding more chemical diversity.
KNIME workflow: remote virtual screening in a cluster
A potential use of Pharmacelera’s nodes is to perform a virtual screening campaign in a workstation or remote cluster. Molecule libraries might be large in size, and, in order to speed up the process this example workflow partitions the dataset in multiple parts and executes it across multiple machines.
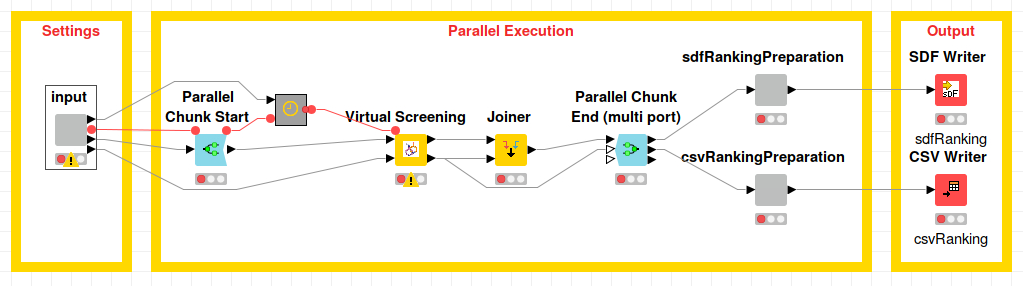
This example Pharmacelera vs MultiServer workflow shows a simple way to deploy virtual screening requiring only the IP addresses of the remote Linux cluster machines, access information, reference molecule and molecule library.
A selection of the most promising molecule candidates is retrieved both in SDF and CSV formats for further postprocessing and analysis. You can download and try out the Pharmacelera_VS_MultiServer example workflow from the KNIME Hub.
Resources
- The Pharmacelera Extensions can be found on the KNIME Hub here.
- Related workflows using these nodes are listed here
About Pharmacelera
Pharmacelera is a trusted KNIME technology partner. PharmScreen nodes enable KNIME users to find more chemical diversity in your virtual screening campaigns. Try out Pharmacelera’s KNIME nodes on the KNIME Hub, and request a demo via the form on the Pharmacelera website.
